




Che cos'è la fibrosi cistica?
Sep 16, 2015
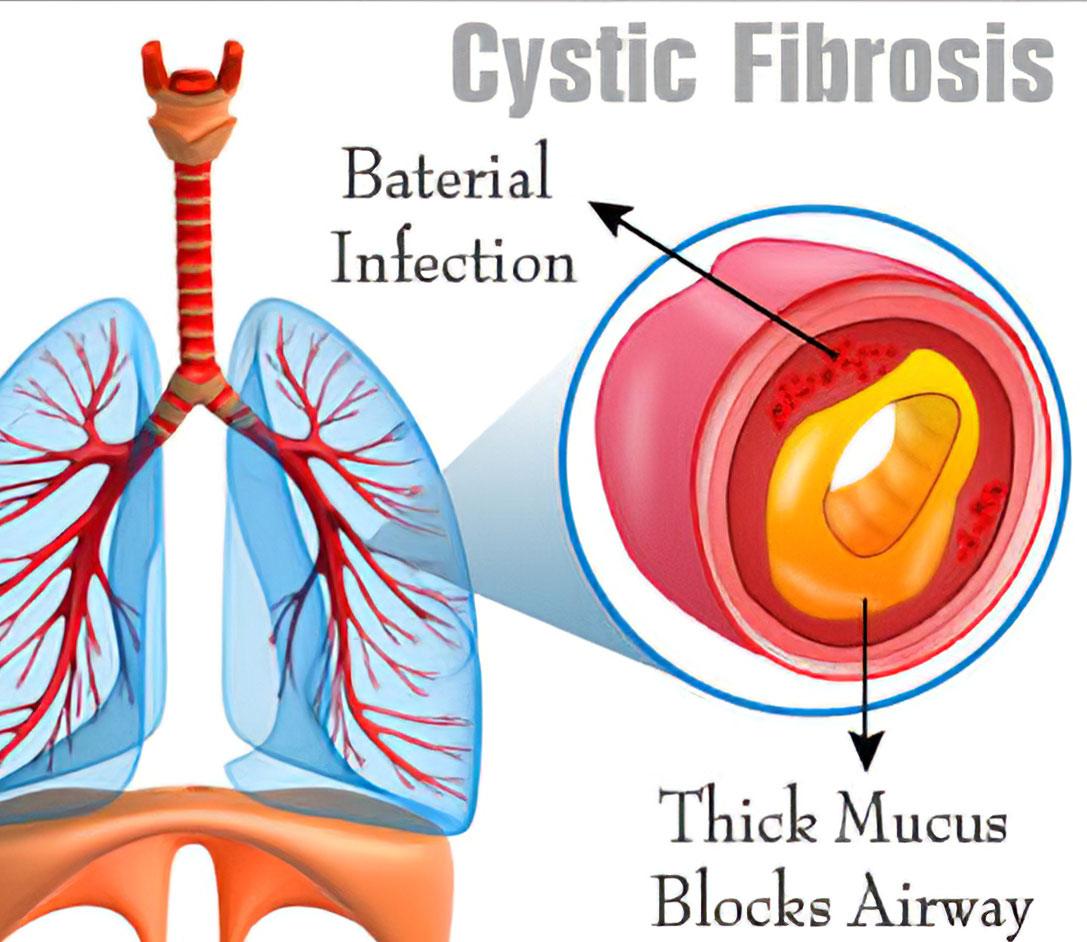
Che cosa è la fibrosi cistica:
La fibrosi cistica è una malattia ereditaria, pericolosa per la vita. Danneggia i polmoni e causa problemi digestivi. Circa 30.000 adulti e bambini americani soffrono di fibrosi cistica.
In passato, non ci si aspettava che le persone affette da fibrosi cistica vivessero oltre l'adolescenza. Oggi, la fibrosi cistica viene diagnosticata prima e trattata in modo più efficace. Di conseguenza, le persone vivono vite più piene fino ai 30, 40 anni e oltre.
La malattia colpisce le cellule che producono muco, sudore, saliva e succhi digestivi. In una persona con fibrosi cistica, queste secrezioni sono spesse e appiccicose. Invece di levigare le superfici su cui si trovano queste secrezioni, ostruiscono tubi, condotti e passaggi, specialmente nei polmoni e nel pancreas. L'insufficienza respiratoria è la complicazione più pericolosa della fibrosi cistica.
Sintomi
Ci sono più di 1.000 mutazioni del gene della fibrosi cistica. La malattia colpisce quasi tutte le ghiandole del corpo che secernono fluidi in un dotto. Colpisce anche molti organi. Alcuni sintomi della fibrosi cistica includono:
Il muco ostruisce le piccole vie aeree dei polmoni, che si infiammano e talvolta si infettano.
Frequenti infezioni dei seni nasali perché i seni si riempiono di muco denso
I linfonodi si gonfiano
Le secrezioni dense possono bloccare i dotti biliari nel fegato. Ciò può portare a gonfiore e dolorabilità (infiammazione) del fegato e alla formazione di cicatrici (cirrosi). La cirrosi può esercitare pressione sulle vene del fegato, facendo sì che le vene nella parte inferiore dell'esofago diventino grandi e fragili. Queste vene possono rompersi e sanguinare.
La cistifellea può bloccarsi
Le secrezioni dense possono bloccare completamente il pancreas impedendo agli enzimi digestivi di raggiungere l'intestino per aiutare a scomporre e assorbire il cibo. Ciò può causare carenze nutrizionali poiché grassi, proteine e vitamine vengono assorbiti male. I bambini con fibrosi cistica crescono più lentamente rispetto agli altri bambini.
Le secrezioni dense che richiedono un intervento chirurgico per essere rimosse in alcuni neonati possono bloccare l'intestino tenue. Tra il 15 e il 20% dei neonati con fibrosi cistica hanno l'ileo da meconio, un grave blocco dell'intestino tenue. Sono anche più inclini alla torsione dell'intestino su se stesso o a un intestino non completamente sviluppato.
Altri possibili sintomi includono:
Un grande appetito che non porta ad aumentare di peso
Pubertà ritardata
Sudorazione eccessiva quando fa caldo o quando si ha la febbre (questa sudorazione espone il paziente a un rischio maggiore di disidratazione).
Frequenti infezioni polmonari o respiratorie e un rischio maggiore di sviluppare un polmone collassato
Feci grasse e voluminose
Perdita di resistenza fisica
Tosse persistente che a volte produce catarro
Pelle dal sapore molto salato o che trasporta cristalli di sale
Respiro sibilante o mancanza di respiro
Uno dei primi segni di fibrosi cistica in un neonato è la lentezza nell'aumento di peso nelle settimane successive alla nascita. A causa della mancanza di enzimi pancreatici per una corretta digestione, il neonato non riceve abbastanza nutrimento per crescere sano. Il neonato potrebbe anche avere lo stomaco dilatato e muscoli piccoli.
Quasi la metà dei bambini con fibrosi cistica viene diagnosticata quando vengono portati dal medico a causa di tosse frequente, respiro sibilante e infezioni delle vie respiratorie. La tosse può essere accompagnata da conati di vomito, vomito e sonno disturbato.
Con il tempo, il torace diventa a forma di botte. La mancanza di ossigeno fa sì che le dita siano più grandi sulla punta. L'area sotto le unghie appare bluastra.
A causa dei problemi digestivi causati dalla fibrosi cistica, possono svilupparsi diverse complicazioni nutrizionali. Tra queste, cecità notturna, rachitismo, anemia (mancanza di ferro nel sangue) e disturbi emorragici. Circa il 15% degli adulti con fibrosi cistica sviluppa diabete che richiede un trattamento con insulina.
La condizione colpisce anche l'apparato riproduttivo. Oltre il 95% degli uomini con fibrosi cistica è sterile. Sebbene molte donne con fibrosi cistica siano in grado di concepire figli, l'impatto della malattia sui loro polmoni e sulla loro salute può rendere piuttosto difficile portare a termine una gravidanza.
Cause e fattori di rischio
Un gene difettoso causa la fibrosi cistica. Il gene controlla la produzione di una proteina che controlla il modo in cui il sale viene trasportato attraverso le membrane che separano le cellule. Le persone con una copia del gene difettoso lo portano ma non hanno sintomi, il che è il caso di oltre 10 milioni di americani. Tuttavia, coloro che ricevono una copia del gene difettoso da entrambi i genitori svilupperanno la fibrosi cistica.
La fibrosi cistica colpisce i caucasici cinque volte più spesso degli afroamericani. È rara nei bambini asiatico-americani. Colpisce in egual misura maschi e femmine.
Diagnosi
La fibrosi cistica è una patologia con cui si nasce. Oltre l'80 percento delle persone affette da fibrosi cistica viene diagnosticata entro i tre anni di età. Alcune forme lievi potrebbero non essere diagnosticate fino a quando la persona non ha 40 o 50 anni.
Per diagnosticare la fibrosi cistica si usa un test del sudore. Semplice e indolore, questo test consiste nell'applicare sulla pelle un farmaco che provoca sudorazione. Per raccogliere il sudore si usa carta da filtro o un tubicino. Avere molto sale nel sudore conferma la fibrosi cistica.
In un neonato, un esame del sangue viene utilizzato per verificare la quantità di un enzima digestivo (tripsina) nel sangue. Un livello elevato mostra la presenza di fibrosi cistica.
I test genetici possono essere utilizzati per confermare una diagnosi di fibrosi cistica in una persona che presenta uno o più sintomi tipici o ha un fratello con fibrosi cistica. Tuttavia, i test genetici possono confermare solo una piccola percentuale degli oltre 1.000 diversi tipi di mutazioni genetiche che possono portare alla fibrosi cistica. I test genetici possono essere eseguiti in fase prenatale tramite prelievo dei villi coriali o amniocentesi.
Poiché la fibrosi cistica colpisce molti organi, potrebbero essere utili altri esami, tra cui:
Analisi di un campione di feci per verificare se i livelli degli enzimi pancreatici sono bassi, in particolare i livelli di tripsina e chimotripsina o se i livelli di grassi sono alti
Esame della glicemia perché se non arriva abbastanza insulina al corpo dal pancreas, la glicemia sarà alta
Test di funzionalità polmonare per verificare se la respirazione è normale
Radiografie del torace, che possono mostrare la presenza di un'infezione o di un danno polmonare
Tomografia computerizzata (TC), che può anche mostrare infezioni o danni polmonari
Trattamento
Non esiste ancora una cura per la fibrosi cistica. Il trattamento è mirato a prevenire complicazioni, come infezioni polmonari o problemi digestivi. Il modo in cui viene trattata la condizione dipende dallo stadio della malattia e dagli organi interessati. È necessario seguire un programma giornaliero per eliminare il muco dai polmoni. Questo può essere fatto:
Fisioterapia toracica in cui la schiena e il torace vengono colpiti vigorosamente per rompere il muco denso nei polmoni
Assumere un farmaco che fluidifica il muco, come Pulmozyme®, per migliorare il funzionamento dei polmoni e prevenire le infezioni polmonari
Utilizzo di un antibiotico per aerosol, come la tobramicina, per prevenire le infezioni polmonari
Utilizzo di azitromicina, un antibiotico efficace per le persone affette da fibrosi cistica i cui polmoni sono cronicamente infetti dal batterio Pseudomonas aeruginos
A causa degli effetti della fibrosi cistica sul sistema digerente di una persona, potrebbe essere necessario assumere sostituti degli enzimi pancreatici a ogni pasto. I pazienti potrebbero anche dover assumere il doppio delle quantità giornaliere raccomandate di vitamine liposolubili (A, D, E e K) in una forma più facile da assorbire.
I bambini con fibrosi cistica hanno bisogno di supporto psicologico e sociale perché potrebbero non essere in grado di partecipare alle normali attività infantili e potrebbero sentirsi isolati. I neonati e i bambini piccoli dovrebbero sottoporsi a vaccinazioni contro la polmonite come parte della loro cura regolare.
Con l'avanzare dell'età, una persona affetta da fibrosi cistica può incorrere in ulteriori problemi di salute, come diabete e osteoporosi.
A un certo punto, potrebbe essere necessario un intervento chirurgico per curare infezioni gravi, sanguinamento nell'esofago, malattia della cistifellea o blocchi intestinali. In alcuni casi, potrebbe essere eseguito un trapianto di fegato o un trapianto di polmone doppio.
La fibrosi cistica è una malattia ereditaria, pericolosa per la vita. Danneggia i polmoni e causa problemi digestivi. Circa 30.000 adulti e bambini americani soffrono di fibrosi cistica.
In passato, non ci si aspettava che le persone affette da fibrosi cistica vivessero oltre l'adolescenza. Oggi, la fibrosi cistica viene diagnosticata prima e trattata in modo più efficace. Di conseguenza, le persone vivono vite più piene fino ai 30, 40 anni e oltre.
La malattia colpisce le cellule che producono muco, sudore, saliva e succhi digestivi. In una persona con fibrosi cistica, queste secrezioni sono spesse e appiccicose. Invece di levigare le superfici su cui si trovano queste secrezioni, ostruiscono tubi, condotti e passaggi, specialmente nei polmoni e nel pancreas. L'insufficienza respiratoria è la complicazione più pericolosa della fibrosi cistica.
Sintomi
Ci sono più di 1.000 mutazioni del gene della fibrosi cistica. La malattia colpisce quasi tutte le ghiandole del corpo che secernono fluidi in un dotto. Colpisce anche molti organi. Alcuni sintomi della fibrosi cistica includono:
Il muco ostruisce le piccole vie aeree dei polmoni, che si infiammano e talvolta si infettano.
Frequenti infezioni dei seni nasali perché i seni si riempiono di muco denso
I linfonodi si gonfiano
Le secrezioni dense possono bloccare i dotti biliari nel fegato. Ciò può portare a gonfiore e dolorabilità (infiammazione) del fegato e alla formazione di cicatrici (cirrosi). La cirrosi può esercitare pressione sulle vene del fegato, facendo sì che le vene nella parte inferiore dell'esofago diventino grandi e fragili. Queste vene possono rompersi e sanguinare.
La cistifellea può bloccarsi
Le secrezioni dense possono bloccare completamente il pancreas impedendo agli enzimi digestivi di raggiungere l'intestino per aiutare a scomporre e assorbire il cibo. Ciò può causare carenze nutrizionali poiché grassi, proteine e vitamine vengono assorbiti male. I bambini con fibrosi cistica crescono più lentamente rispetto agli altri bambini.
Le secrezioni dense che richiedono un intervento chirurgico per essere rimosse in alcuni neonati possono bloccare l'intestino tenue. Tra il 15 e il 20% dei neonati con fibrosi cistica hanno l'ileo da meconio, un grave blocco dell'intestino tenue. Sono anche più inclini alla torsione dell'intestino su se stesso o a un intestino non completamente sviluppato.
Altri possibili sintomi includono:
Un grande appetito che non porta ad aumentare di peso
Pubertà ritardata
Sudorazione eccessiva quando fa caldo o quando si ha la febbre (questa sudorazione espone il paziente a un rischio maggiore di disidratazione).
Frequenti infezioni polmonari o respiratorie e un rischio maggiore di sviluppare un polmone collassato
Feci grasse e voluminose
Perdita di resistenza fisica
Tosse persistente che a volte produce catarro
Pelle dal sapore molto salato o che trasporta cristalli di sale
Respiro sibilante o mancanza di respiro
Uno dei primi segni di fibrosi cistica in un neonato è la lentezza nell'aumento di peso nelle settimane successive alla nascita. A causa della mancanza di enzimi pancreatici per una corretta digestione, il neonato non riceve abbastanza nutrimento per crescere sano. Il neonato potrebbe anche avere lo stomaco dilatato e muscoli piccoli.
Quasi la metà dei bambini con fibrosi cistica viene diagnosticata quando vengono portati dal medico a causa di tosse frequente, respiro sibilante e infezioni delle vie respiratorie. La tosse può essere accompagnata da conati di vomito, vomito e sonno disturbato.
Con il tempo, il torace diventa a forma di botte. La mancanza di ossigeno fa sì che le dita siano più grandi sulla punta. L'area sotto le unghie appare bluastra.
A causa dei problemi digestivi causati dalla fibrosi cistica, possono svilupparsi diverse complicazioni nutrizionali. Tra queste, cecità notturna, rachitismo, anemia (mancanza di ferro nel sangue) e disturbi emorragici. Circa il 15% degli adulti con fibrosi cistica sviluppa diabete che richiede un trattamento con insulina.
La condizione colpisce anche l'apparato riproduttivo. Oltre il 95% degli uomini con fibrosi cistica è sterile. Sebbene molte donne con fibrosi cistica siano in grado di concepire figli, l'impatto della malattia sui loro polmoni e sulla loro salute può rendere piuttosto difficile portare a termine una gravidanza.
Cause e fattori di rischio
Un gene difettoso causa la fibrosi cistica. Il gene controlla la produzione di una proteina che controlla il modo in cui il sale viene trasportato attraverso le membrane che separano le cellule. Le persone con una copia del gene difettoso lo portano ma non hanno sintomi, il che è il caso di oltre 10 milioni di americani. Tuttavia, coloro che ricevono una copia del gene difettoso da entrambi i genitori svilupperanno la fibrosi cistica.
La fibrosi cistica colpisce i caucasici cinque volte più spesso degli afroamericani. È rara nei bambini asiatico-americani. Colpisce in egual misura maschi e femmine.
Diagnosi
La fibrosi cistica è una patologia con cui si nasce. Oltre l'80 percento delle persone affette da fibrosi cistica viene diagnosticata entro i tre anni di età. Alcune forme lievi potrebbero non essere diagnosticate fino a quando la persona non ha 40 o 50 anni.
Per diagnosticare la fibrosi cistica si usa un test del sudore. Semplice e indolore, questo test consiste nell'applicare sulla pelle un farmaco che provoca sudorazione. Per raccogliere il sudore si usa carta da filtro o un tubicino. Avere molto sale nel sudore conferma la fibrosi cistica.
In un neonato, un esame del sangue viene utilizzato per verificare la quantità di un enzima digestivo (tripsina) nel sangue. Un livello elevato mostra la presenza di fibrosi cistica.
I test genetici possono essere utilizzati per confermare una diagnosi di fibrosi cistica in una persona che presenta uno o più sintomi tipici o ha un fratello con fibrosi cistica. Tuttavia, i test genetici possono confermare solo una piccola percentuale degli oltre 1.000 diversi tipi di mutazioni genetiche che possono portare alla fibrosi cistica. I test genetici possono essere eseguiti in fase prenatale tramite prelievo dei villi coriali o amniocentesi.
Poiché la fibrosi cistica colpisce molti organi, potrebbero essere utili altri esami, tra cui:
Analisi di un campione di feci per verificare se i livelli degli enzimi pancreatici sono bassi, in particolare i livelli di tripsina e chimotripsina o se i livelli di grassi sono alti
Esame della glicemia perché se non arriva abbastanza insulina al corpo dal pancreas, la glicemia sarà alta
Test di funzionalità polmonare per verificare se la respirazione è normale
Radiografie del torace, che possono mostrare la presenza di un'infezione o di un danno polmonare
Tomografia computerizzata (TC), che può anche mostrare infezioni o danni polmonari
Trattamento
Non esiste ancora una cura per la fibrosi cistica. Il trattamento è mirato a prevenire complicazioni, come infezioni polmonari o problemi digestivi. Il modo in cui viene trattata la condizione dipende dallo stadio della malattia e dagli organi interessati. È necessario seguire un programma giornaliero per eliminare il muco dai polmoni. Questo può essere fatto:
Fisioterapia toracica in cui la schiena e il torace vengono colpiti vigorosamente per rompere il muco denso nei polmoni
Assumere un farmaco che fluidifica il muco, come Pulmozyme®, per migliorare il funzionamento dei polmoni e prevenire le infezioni polmonari
Utilizzo di un antibiotico per aerosol, come la tobramicina, per prevenire le infezioni polmonari
Utilizzo di azitromicina, un antibiotico efficace per le persone affette da fibrosi cistica i cui polmoni sono cronicamente infetti dal batterio Pseudomonas aeruginos
A causa degli effetti della fibrosi cistica sul sistema digerente di una persona, potrebbe essere necessario assumere sostituti degli enzimi pancreatici a ogni pasto. I pazienti potrebbero anche dover assumere il doppio delle quantità giornaliere raccomandate di vitamine liposolubili (A, D, E e K) in una forma più facile da assorbire.
I bambini con fibrosi cistica hanno bisogno di supporto psicologico e sociale perché potrebbero non essere in grado di partecipare alle normali attività infantili e potrebbero sentirsi isolati. I neonati e i bambini piccoli dovrebbero sottoporsi a vaccinazioni contro la polmonite come parte della loro cura regolare.
Con l'avanzare dell'età, una persona affetta da fibrosi cistica può incorrere in ulteriori problemi di salute, come diabete e osteoporosi.
A un certo punto, potrebbe essere necessario un intervento chirurgico per curare infezioni gravi, sanguinamento nell'esofago, malattia della cistifellea o blocchi intestinali. In alcuni casi, potrebbe essere eseguito un trapianto di fegato o un trapianto di polmone doppio.