




What is Cystic Fibrosis?
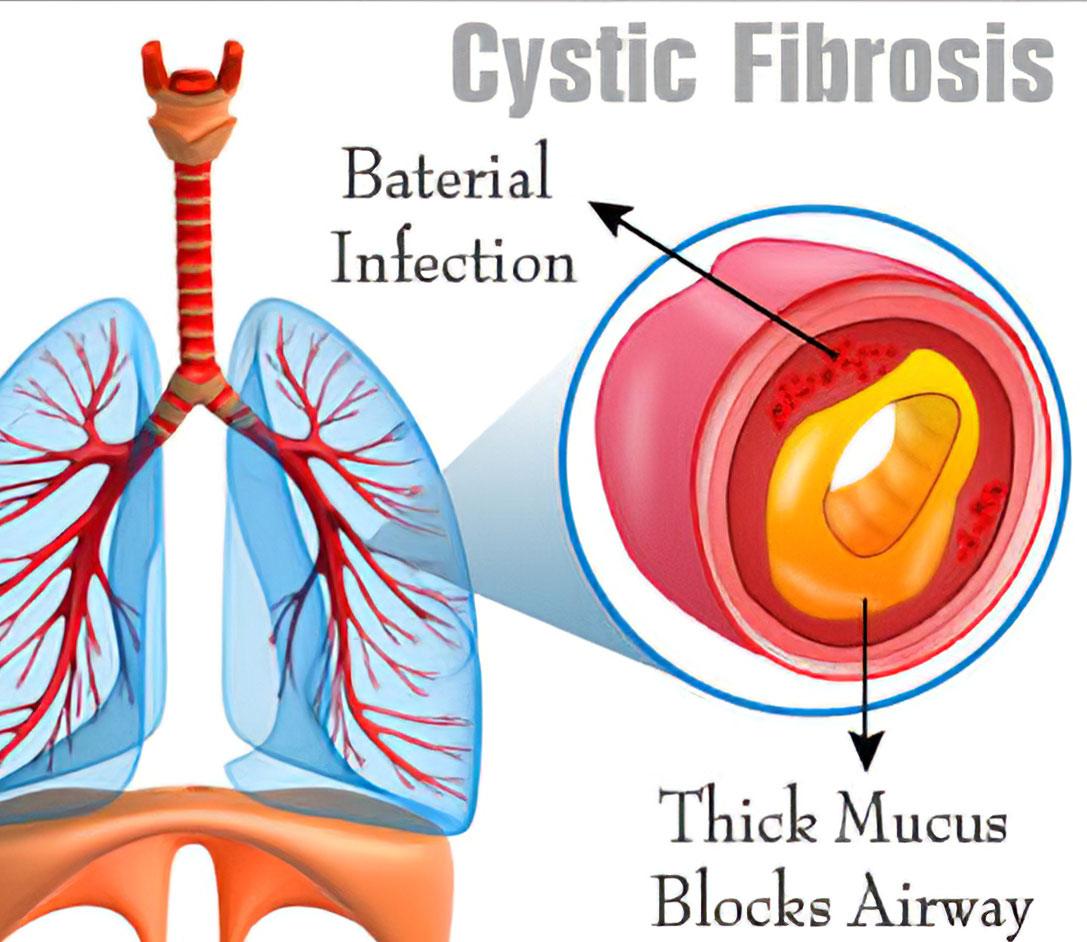
What is Cystic Fibrosis
Cystic fibrosis is an inherited, life threatening disorder. It damages the lungs and causes digestive problems. About 30,000 American adults and children have cystic fibrosis.
In the past, people with cystic fibrosis were not expected to live beyond their teens. Today, cystic fibrosis is diagnosed earlier and treated more effectively. As a result, people live fuller lives into their 30s, 40s and beyond.
The disease affects the cells that produce mucus, sweat, saliva and digestive juices. In a person with cystic fibrosis, these secretions are thick and sticky. Instead of smoothing the surfaces that these secretions are on, they plug up tubes, ducts and passageways, especially in the lungs and pancreas. Respiratory failure is the most dangerous complication of cystic fibrosis.

Cystic Fibrosis Symptoms
There are more than 1,000 mutations of the gene for cystic fibrosis. The disease affects nearly all the body's glands that secrete fluids into a duct. It also affects many organs. Some symptoms of cystic fibrosis include:
- Mucus blocks the small airways of the lungs, which become inflamed and sometimes infected.
- Frequent sinus infections because the sinuses fill with thickened mucus
- The lymph nodes swell
- Thick secretions can block the bile ducts in the liver, leading to swelling, inflammation, cirrhosis, and potentially rupturing veins in the esophagus.
- The gall bladder can become blocked
- Thick secretions can block the pancreas entirely, preventing digestive enzymes from reaching the intestines, causing nutritional deficiencies.
- Thick secretions that require surgery to remove in some newborns can block the small intestine (meconium ileus). 15–20% of newborns with cystic fibrosis have this blockage.
Other Possible Cystic Fibrosis Symptoms
- A big appetite that does not lead to weight gain
- Delayed puberty
- Excessive sweating in hot weather or during a fever (increasing dehydration risk)
- Frequent lung or respiratory infections and a greater risk of collapsed lung
- Greasy, bulky stools
- Loss of physical endurance
- Persistent coughing that sometimes produces phlegm
- Skin that tastes very salty or carries salt crystals
- Wheezing or shortness of breath
One of the first signs in a baby is slow weight gain after birth due to lack of pancreatic enzymes. Nearly half the children with cystic fibrosis are diagnosed because of frequent coughing, wheezing, and respiratory tract infections.
Over time, the chest becomes barrel-shaped, fingertips enlarge, and the area under the nails can appear bluish. Nutritional complications can include night blindness, rickets, anemia, and bleeding disorders. About 15% of adults develop diabetes requiring insulin. More than 95% of men are sterile, and women may have difficulty carrying a pregnancy to term.

Causes and Risk Factors
A defective gene causes cystic fibrosis, affecting salt transport across cell membranes. People with one copy are carriers without symptoms. Those who inherit two defective copies develop the disease. It affects Caucasians more frequently than African Americans, is rare in Asian-American children, and affects both sexes equally.
Diagnosis
Over 80% of cases are diagnosed by age three. Tests include:
- Sweat test: Measures salt levels in sweat
- Newborn blood test: Measures digestive enzyme trypsin
- Genetic testing: Confirms certain mutations
- Stool analysis for enzyme or fat levels
- Blood glucose test
- Lung function tests
- Chest X-rays
- CT scans
Treatment
There is no cure yet. Treatment focuses on preventing complications such as lung infections or digestive problems.
Lung care may include:
- Chest physical therapy
- Mucus-thinning drugs (e.g., Pulmozyme®)
- Aerosol antibiotics (e.g., tobramycin)
- Azithromycin for chronic infections
Digestive care may include:
- Pancreatic enzyme replacements with each meal
- Double the recommended daily amounts of fat-soluble vitamins (A, D, E, K)
Children need psychological and social support. Immunizations against pneumonia are recommended. Additional complications like diabetes and osteoporosis may develop. In severe cases, surgery, liver transplant, or double lung transplant may be necessary.

How Physiologic Instruments’ Ussing Chamber Systems Advance Cystic Fibrosis Research
Cystic fibrosis (CF) is a life-threatening genetic disorder caused by mutations in the CFTR gene, which impair the movement of chloride and bicarbonate ions across epithelial cells. This disruption leads to thick, sticky mucus in the lungs, pancreas, and other organs, driving the severe respiratory and digestive complications associated with the disease.
While there is no cure yet, scientific advances in understanding and treating CF have been accelerated by precise laboratory tools — including the Ussing Chamber Systems from Physiologic Instruments.
Measuring CFTR Function Directly
Ussing Chambers allow researchers to mount live epithelial tissue or cultured cell layers between two fluid-filled chambers, simulating the inside and outside of the body. By measuring short-circuit current (Isc) and transepithelial resistance (TER), scientists can detect how well chloride and bicarbonate ions move across the cell layer — a direct readout of CFTR function.
Testing Potential Therapies
Many modern CF drugs, such as CFTR modulators, aim to correct or enhance ion transport. Using an Ussing Chamber, researchers can:
-
Establish a baseline of CFTR activity in diseased tissue
-
Apply new drug candidates
-
Quantify improvements in ion transport after treatment
This approach confirms whether a therapy is effectively restoring CFTR function.
Driving Personalized Medicine
With patient-derived organoids (miniature versions of tissues grown from stem cells), Ussing Chambers can test how an individual’s cells respond to different drug combinations. This can help clinicians and researchers tailor treatments to a person’s specific genetic mutation.
Impact on the Future of CF Care
Data from Ussing Chamber experiments supports drug development, regulatory submissions, and clinical decision-making, bringing promising treatments to patients faster. By enabling precise, reproducible measurements of epithelial ion transport, Physiologic Instruments’ systems play an important role in the global effort to improve — and one day cure — cystic fibrosis.