




Exon-skipping antisense oligonucleotides for cystic fibrosis therapy
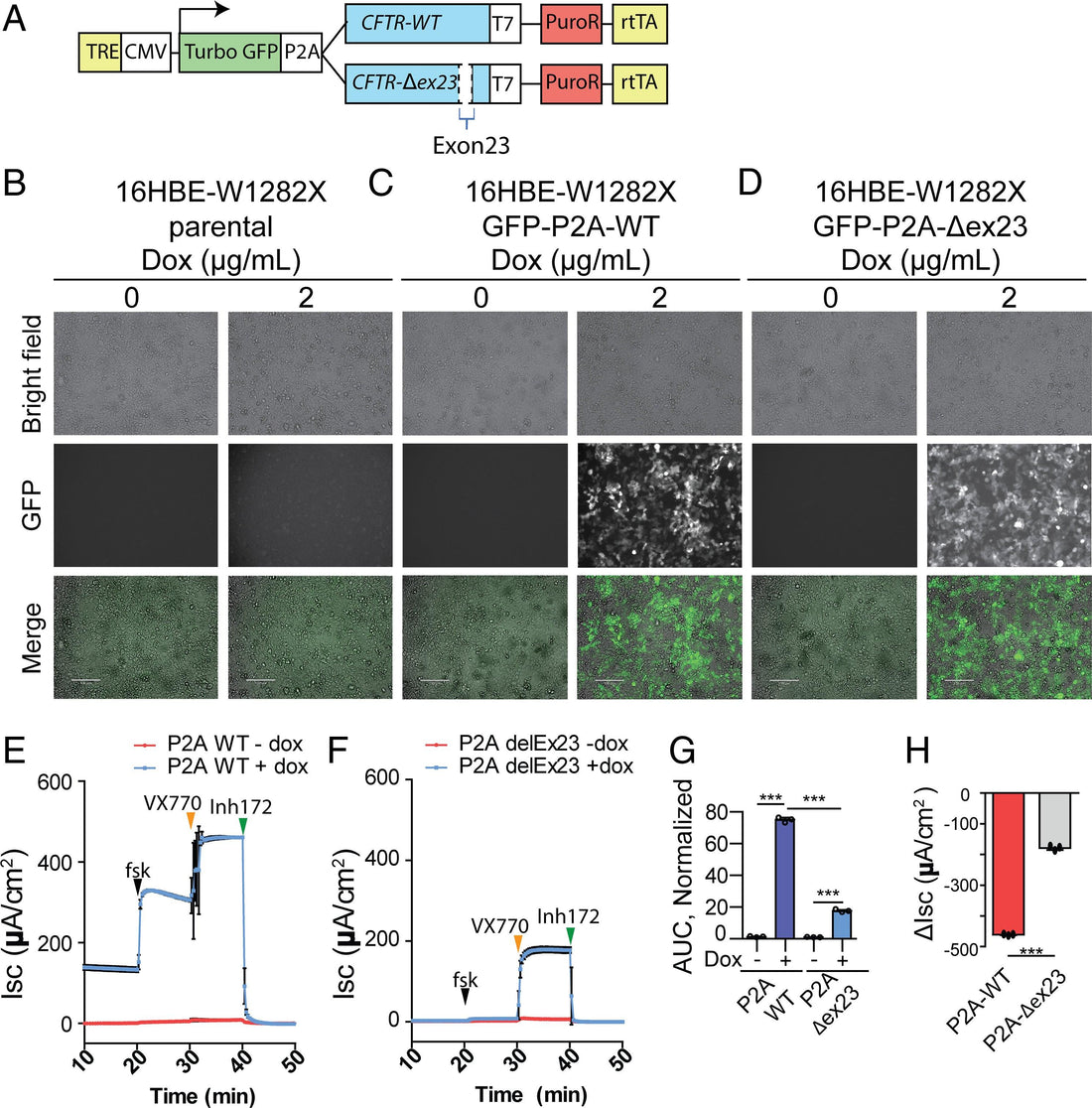
Mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene cause cystic fibrosis (CF), and the CFTR-W1282X nonsense mutation causes a severe form of CF. Although Trikafta and other CFTR-modulation therapies benefit most CF patients, targeted therapy for patients with the W1282X mutation is lacking. The CFTR-W1282X protein has residual activity but is expressed at a very low level due to nonsense-mediated messenger RNA (mRNA) decay (NMD). NMD-suppression therapy and read-through therapy are actively being researched for CFTR nonsense mutants. NMD suppression could increase the mutant CFTR mRNA, and read-through therapies may increase the levels of full-length CFTR protein. However, these approaches have limitations and potential side effects: because the NMD machinery also regulates the expression of many normal mRNAs, broad inhibition of the pathway is not desirable, and read-through drugs are inefficient partly because the mutant mRNA template is subject to NMD. To bypass these issues, we pursued an exon-skipping antisense oligonucleotide (ASO) strategy to achieve gene-specific NMD evasion. A cocktail of two splice-site–targeting ASOs induced the expression of CFTR mRNA without the premature-termination-codon–containing exon 23 (CFTR-Δex23), which is an in-frame exon. Treatment of human bronchial epithelial cells with this cocktail of ASOs that target the splice sites flanking exon 23 results in efficient skipping of exon 23 and an increase in CFTR-Δex23 protein. The splice-switching ASO cocktail increases the CFTR-mediated chloride current in human bronchial epithelial cells. Our results set the stage for developing an allele-specific therapy for CF caused by the W1282X mutation.
"